#Industry News
Is glioma classification too complicated? After reading this article, learn about the “brain killer”—glioma
Is glioma classification too complicated? After reading this article, learn about the “brain killer”—glioma
Glioma overview
Glioma is a tumor originating from glial cells in the brain and is the most common primary intracranial tumor. The pathogenesis of glioma is unknown, but two risk factors currently identified are exposure to high doses of ionizing radiation and high-penetrance genetic mutations associated with rare syndromes. In addition, carcinogenic factors such as nitrite foods, viral or bacterial infections may also be involved in the occurrence of brain glioma. The annual incidence rate of brain glioma in my country is 5/100,000 to 8/100,000, and the 5-year mortality rate is second only to pancreatic cancer and lung cancer among systemic tumors [1]. Gliomas occur in all ages, are more common in adults, and are more common in men than women. According to the fifth edition of the WHO Classification of Central Nervous System Tumors (WHO CNS5), the standards of care and prognosis for gliomas differ significantly. Generally speaking, gliomas are divided into localized gliomas and diffuse gliomas. The former is benign and can be cured after complete surgical resection, while the latter is more malignant and cannot be cured after surgical resection alone. The new classification divides gliomas into grades 1 to 4. Grades 1 and 2 are low-grade gliomas (LGG), and grades 3 and 4 are high-grade gliomas (HGG), It also integrated the histological characteristics and molecular phenotypes of tumors, proposed new tumor classification criteria, and focused on promoting the application of molecular diagnosis in the classification of central nervous system tumors. This classification is an important basis for the current diagnosis and grading of brain glioma. The new classification divides brain gliomas into five groups based on histological and molecular pathology characteristics: adult diffuse glioma, pediatric diffuse LGG, pediatric diffuse HGG, and localized astrocytic glioma. tumours, ependymal tumors [1].
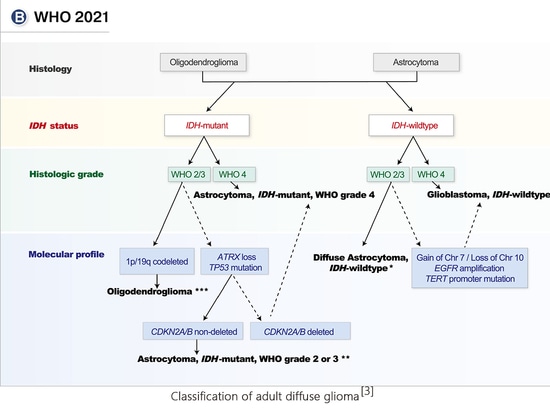
adult diffuse glioma
Adult diffuse glioma patients are divided into 3 types based on histopathology, mutation spectrum, and copy number changes: astrocytoma, IDH mutant type; oligodendroglioma, IDH mutant, 1p/19q codeletion type ;Glioblastoma, IDH wild type.
Astrocytoma, IDH mutant type, needs to meet the requirements of both histological morphology and molecular characteristics. Patients of this type are divided into 3 levels, namely CNS WHO levels 2-4. The presence of a homozygous deletion of CDKN2A/B can be assessed as grade 4, regardless of the absence of high-grade features on histology.
If IDH-mutated diffuse glioma is accompanied by 1p/19q codeletion, it is diagnosed as oligodendroglioma, IDH mutation and 1p/19q codeletion type. It can be diagnosed as grade 2 or 3 based on histopathological characteristics. 1p/19q codeletion is closely associated with IDH mutations, so true full-arm 1p/19q codeletion is extremely rare in IDH wild-type tumors.
Invasive astrocytomas lack the histological features of glioblastoma (necrosis and/or microvascular proliferation) but possess one or more molecular hallmarks of glioblastoma, including the following: EGFR amplification , amplification of chromosome 7 and deletion of chromosome 10; TERT promoter mutation. In this case, the tumor could still be diagnosed as glioblastoma, IDH wild-type, WHO grade 4. These tumors have similar clinical outcomes to typical histological grade 4 IDH wild-type glioblastoma and can therefore be treated accordingly.
childhood diffuse glioma
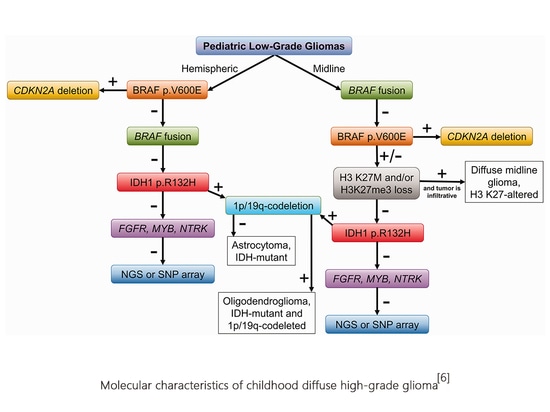
The pathogenesis, treatment targets, and prognosis of diffuse glioma in children are very different from those in adults. The 2021 WHO classification divides childhood gliomas into two categories, namely childhood diffuse low-grade glioma and childhood diffuse high-grade glioma.
Almost all pediatric diffuse low-grade gliomas are driven by changes in the up-regulation of the RAS/MAPK pathway. Their histological appearance is diffuse low-grade glioma, which mainly occurs in children but can also be seen in adults. Usually Have a history of epilepsy. Molecular variations are divided into two categories: MYB or MYBL1 variants and MAPK pathway variants.
1) MYB or MYBL1 variants: MYB or MYBLI variants include gene copy number variations and gene fusions (MYB partner genes include QKI, ESR1, MMP16, MAML2, PCDHGA1, etc., MYBL1 partner genes include RAD51B, MAML2, ZFHX4, TOX, etc.) The new tumor classification combines histological morphology and molecular characteristics to divide MYB or MYBL1 variant tumors into two types: diffuse astrocytoma, MYB or MYBL1 variant and angiocentric glioma (MYB-QKI fusion is common).
2) Gene mutations related to MAPK pathway in glioma include NF1, BRAF, FGFR1, CRAF, NTRK, PTPN11, ROS1, etc. Molecular alterations in polymorphic low-grade neuroepithelial neoplasia of the young (PLNTY) include BRAF V600E, FGFR3-TACC3 fusion, FGFR2-CTNNA3 fusion, and FGFR2-KIAA1598 fusion. In diffuse low-grade gliomas, common molecular alterations in MAPK pathway variants include FGFR1 tyrosine kinase domain (TKD) duplications, FGFR1 mutations, FGFR1 fusions, and BRAF V600E mutations, BRAF fusions, and BRAF insertion mutations [4] .
Diffuse high-grade glioma in children is a rare but biologically diverse tumor. Pediatric diffuse high-grade glioma is a typical diffuse infiltrative glioma (the most common astrocytoma in nature). It is further divided into diffuse midline glioma based on its histological morphology and molecular characteristics. , H3K27 mutant, diffuse hemispheric glioma, H3G34 mutant, diffuse childhood high-grade glioma, H3 wild type and IDH wild type, infantile hemispheric glioma, a total of 4 types, all of which are CNS WHO grade 4.
Diffuse midline glioma, H3K27 variant, occurs in the midline of the central nervous system. It is divided into 3 subtypes based on molecular variation, namely H3 K27 mutant type (including H3 K27M and H3K27I), EGFR mutant type (mostly involving the thalamus), and H3 wild type with EZHIP overexpression. H3 K27 mutations occur in H3.3 (encoding gene H3F3A) and H3.1 (encoding gene HIST1H3B/C). Among them, the mutation rate of H3F3A is about 3 times that of HIST1H3B/C, and the prognosis is worse. Molecular variations such as TP53 mutation, ACVR1 mutation, PPM1D mutation and PDGFRA amplification are common molecular genetic features of H3 K27 mutant diffuse midline glioma [6].
Another tumor carrying histone H3 mutations is diffuse hemispheric glioma, H3 G34 mutant type, which mainly occurs in the cerebral hemisphere, showing that glycine (Gly) at position 34 of histone H3.3 is replaced by arginine (Arg) or Valine (Val) substitution missense mutation (H3.3 G34R/V). H3.3G34 mutation in glioma mainly occurs in the H3F3A gene, often accompanied by ATRX mutation and TP53 mutation.
H3 wild type and IDH wild type: diffuse childhood high-grade glioma. H3 wild type and IDH wild type tend to occur in children and young adults. They have histological characteristics of high-grade tumors, but the molecular pathological characteristics are IDH wild type, group Protein H3 wild type. According to DNA methylation characteristics, it can be divided into childhood high-grade glioma RTK1 type, childhood high-grade glioma RTK2 type, and childhood high-grade glioma MYCN type.
Infantile hemispheric glioma occurs in infancy. The lesions are located in one cerebral hemisphere and have different histological and morphological characteristics. More than one-third of the tumors have receptor tyrosine kinase (RTK) changes, especially ALK and ROS1. , fusion of NTRK and MET genes [3].
localized astrocytic glioma
Localized astrocytic glioma has clear borders, localized growth is the main feature, and the prognosis is good. The most common molecular mutation of pilocytic astrocytoma is KIAA1549-BRAF fusion (>70%), and pleomorphic xanthoma type The most common astrocytomas are BRAF V600E mutations, and other mutations include homozygous deletion of CDKN2A/B and gains of chromosomes 3 and 7. The incidence of subependymal giant cell astrocytoma is closely related to multiple sclerosis, so it is often accompanied by TSC1 and TSC2 mutations. The most common molecular mutation in chordoid glioma is PRKCA D463H mutation, and some tumors can also carry BRAF V600E mutation. High-grade astrocytomas with pilocytoid features are associated with high frequency of MAPK pathway gene mutations. Tumors with typical astroblastoma histological morphology can be diagnosed as astroblastoma if they carry MN1 mutations (MN1-BEND2 fusion and MN1-CXXC5 fusion, large loss of heterozygosity on autosomal chromosome 22q and X chromosome) Tumor MNI is abnormal [4].
Ependymoma
Supratentorial ependymoma is mainly characterized by fusion genes and can be divided into ZFTA fusion-positive type and YAP1 fusion-positive type. The proportion of supratentorial ependymomas with non-ZFTA and non-YAP1 fusions is lower. Posterior fossa (PF) ependymoma shows characteristic DNA methylation profile changes and can be divided into PFA group and PFB group; PFA group ependymomas mainly occur in infants and young children. Most have anaplastic characteristics , its prognosis is poor, histone H3K27me3 expression is lost, EZHIP is overexpressed, and the genome is relatively stable; ependymoma in the PFB group mainly occurs in older children or adults, and its prognosis is relatively good, with normal H3 K27me3 expression. One type of spinal cord ependymoma is characterized by MYCN gene amplification and has strong invasiveness and metastatic ability, and its prognosis is poor.
If molecular testing has not been performed or molecular testing is not available, the suffix NOS can be used to indicate it. If molecular testing has been performed, but the results are not sufficient for diagnosis or classification, the suffix NEC can be used.
Glioma gene testing
In order to meet the clinical needs of patients with brain glioma, SpaceGen has launched a glioma gene testing service, which combines with the second-generation sequencing platform to detect point mutations, indels, fusions, copy number variations, chromosomal variations, methylation, etc. The mutated form provides 54 genes + MSI gene testing to help individualized and precise diagnosis and treatment of glioma patients!
Project parameters
Detection significance
1. Auxiliary classification: The diagnosis of brain glioma requires specimens to be obtained through tumor resection or biopsy, and tissue and molecular pathology examination to determine the pathological grade and molecular subtype. At present, the main molecular pathological markers include: IDH1/2 mutation, TERT mutation, H3F3A mutation, BRAF mutation, etc.
2. Prognostic assessment: Different molecular subtypes and markers may have different prognostic characteristics. Among gliomas of all grades, patients with IDH mutation have a better prognosis than IDH wild-type. The impact of IDH mutation status on the prognosis of glioma is considered to be better than histological grade.
3. Guide treatment: Molecular testing for glioblastoma is encouraged. If driver mutations (such as BRAF V600E mutations or NTRK fusions) are detected, targeted therapy can be performed, and patients may have more treatment options in clinical trials.
Advantages and features
Comprehensive testing: Comprehensive coverage of the important clinical significance of genetic testing in the diagnosis and treatment of glioma, including targeted medication, immune medication, efficacy prediction of chemotherapy drugs, toxic side tolerance and drug resistance assessment, genetic determination, etc.
High sensitivity: Probe capture + NGS are used for deep sequencing to reduce missed detections and maximize the possibility of drug use.
Rigorous testing: From sample collection to report issuance, strict quality control standards are implemented throughout the entire process.
Technical expertise: The independently developed automated analysis system interprets the report, and is equipped with a professional medical team to review the report, truly achieving comprehensive and accurate interpretation and providing scientific basis for individualized medication for patients.
References
1 Guidelines for Diagnosis and Treatment of Glioma (2022 Edition)
2 World J Clin Oncol. 2022 Jul 24;13(7):567-576.
3.Cancer. 2022 Jan 1;128(1):47-58.
4. Interpretation of molecular diagnostic indicators in the 2021 World Health Organization Classification of Central Nervous System Tumors (Fifth Edition)
5.Pediatr Dev Pathol. 2022 Jan-Feb;25(1):46-58.
6.NCCN Guidelines Version 1.2023Central Nervous System Cancers
Statement: This article is for sharing only and does not represent the position of the platform. If there are copyright and other issues involved, please contact us as soon as possible and we will correct it as soon as possible. Thank you!