#Industry News
Presentation of tumor neoantigens
Presentation of tumor neoantigens
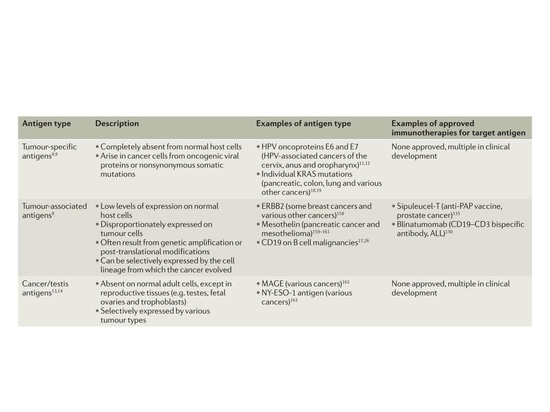
Classification of tumor antigens
The latest advances in genome sequencing indicate that during initiation and development, cancer acquires tens of thousands of different somatic cell mutations. Most of these mutations do not confer inherent growth advantages (passenger mutations) and are often a result of genomic instability within the tumor.
A small subset of cancer mutations disrupt normal cellular regulation and contribute to driving cancer growth and resistance to targeted therapies (driver mutations). So far, approximately 140 genes have been identified that can drive tumorigenesis. However, both driver and passenger mutations can alter the amino acid coding sequence, collectively referred to as synonymous mutations, leading to mutated proteins not expressed in normal cells. These aberrant protein sequences are processed into short peptides and bind to the major histocompatibility complex (MHC; also known as human leukocyte antigen HLA in humans), making them recognizable by T cells as foreign antigens [1].
Due to their selective expression in tumors, tumor-specific antigens (TSAs) generated by synonymous mutations and other genetic alterations are referred to as neoantigens. In human tumor subgroups with a viral etiology, such as Merkel cell carcinoma (MCC) associated with Merkel cell polyomavirus (MCPyV) and cervical, oral, and other site-specific cancers associated with human papillomavirus (HPV), proteins encoded by the viral open reading frame represent another type of neoantigen. In addition to TSAs, there are two major classes of tumor antigens. Tumor-associated antigens (TAAs) are overexpressed in malignant cells but also expressed at low levels in normal cells. Cancer/testis antigens (CTAs) are expressed by various tumor types and reproductive tissues (e.g., testes, fetal ovaries, and nourishing layers) but have limited expression in other normal adult tissues and are usually absent in normal reproductive cells because these tissues do not express MHC class molecules. Neoantigens can occur at the genomic level through single nucleotide variations (SNVs), base insertions, and gene fusions, at the transcriptional level through selective splicing, polyadenylation (pA), RNA editing, and so-called non-coding regions, and at the protein level through disrupted translation and post-translational modifications.
▲Classification of tumor antigens[1]
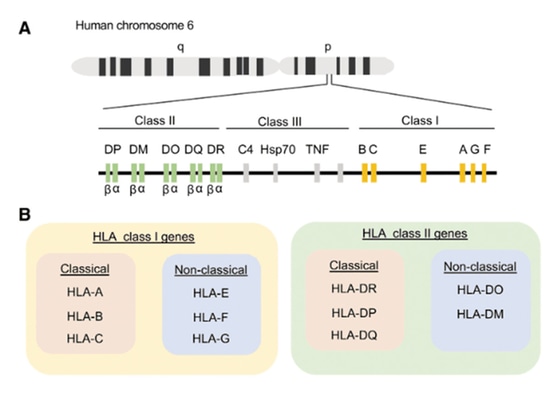
Classification of HLA
The activation of T cells depends on the simultaneous recognition of foreign peptide fragments and self-MHC molecules, a phenomenon known as MHC restriction. CD8+ T cells are restricted by MHC-I, while CD4+ T cells are restricted by MHC-II. The major histocompatibility complex (MHC), referred to as human leukocyte antigen (HLA) in humans, is encoded on chromosome 6 of the human genome. It is a highly polymorphic gene complex that encodes cell surface molecules specialized in presenting and recognizing self and non-self peptides. HLA is divided into three classes based on function and structure: HLA-I, HLA-II, and HLA-III. HLA class molecules are expressed on the surface of nucleated cells, excluding germ cells and some neuronal cells. Unlike HLA-I class molecules, HLA-II class molecules are typically found on professional antigen-presenting cells (APCs) such as B cells, macrophages, dendritic cells, Langerhans cells, thymic epithelium, and activated (rather than resting) T cells. The structure and function of HLA-III class molecules are not well understood, but they are known to participate in the inflammatory process without directly binding to antigens.
HLA is further divided into classical and non-classical genes. Classical HLA-I genes include HLA-A, HLA-B, and HLA-C, while non-classical alleles include HLA-E, HLA-F, and HLA-G. Classical HLA-II genes include HLA-DR, HLA-DP, and HLA-DQ, while non-classical alleles include HLA-DO and HLA-DM. Human CD8+ T cells recognize peptides presented by classical HLA-A and HLA-B and, to a lesser extent, by HLA-C. Human CD4+ T cells recognize peptides presented by HLA-DR, HLA-DQ, and HLA-DP. HLA-I consists of three extracellular domains (α1, α2, and α3) non-covalently linked to a β2-microglobulin molecule. HLA-II is a heterodimer composed of an α and a β chain. The extracellular domains of HLA form an antigen-binding cleft, consisting of two α-helices surrounding anti-parallel β-folded sheets. This creates a platform capable of accommodating a short amino acid (aa) segment called a peptide. These peptides bind to the bottom of the binding groove through interactions with specific amino acids (called anchor residues). Due to the closed structure of the HLA-I binding cleft, it typically binds small peptides of 8-10 amino acids, while HLA-II can bind longer peptides exceeding 11 amino acids.
▲Classification of diffuse gliomas in adults[2]
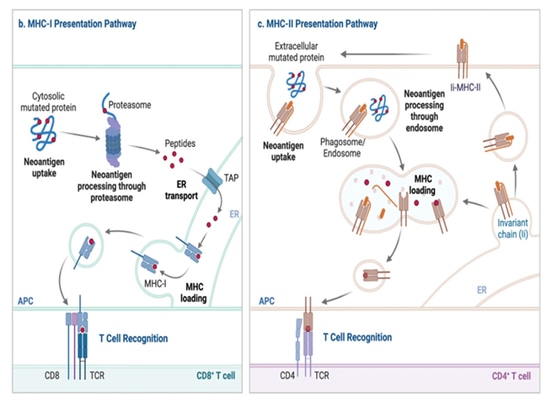
Presentation of neoantigens
The presentation of MHC-I proteins primarily originates from peptides inside the cell. During homeostasis, cellular proteins are degraded by proteasomes, generating small peptides. Proteasomes are multiprotein complexes that degrade proteins into small peptide fragments. During viral infection, the action of interferons induces the formation of an alternative proteasome complex called the immunoproteasome, which enhances the generation of MHC-I-presented peptides. Consequently, when synthesizing viral proteins, they are targeted for degradation by the immunoproteasome, either as fully folded proteins or as defective ribosomal products. This results in the production of small peptide fragments derived from the infecting virus, which can be further modified by cytoplasmic aminopeptidases.
These peptide fragments are then transported into the endoplasmic reticulum (ER) by a protein transport mechanism known as the antigen processing related transport protein (TAP). In the ER, the peptides undergo further modification by endoplasmic reticulum aminopeptidase 1 (ERAP1) if they are too long for MHC-I binding after translocation through TAP. In the ER, empty MHC-I molecules associate with peptide-loading complexes (PLC), including chaperone proteins such as Tapasin and Calnexin. PLC maintains the empty MHC-I in a peptide-receptive conformation, and when peptides are translocated by TAP, this conformation facilitates peptide binding. TAP is also a part of PLC. Peptide binding stabilizes the MHC-I protein, releasing it from the endoplasmic reticulum quality control partners, and it is transported to the cell surface through the Golgi apparatus.
This process allows CD8+ T cells to survey for signs of infection or malignancy within the cellular protein repertoire. However, in specific subsets of dendritic cells, an alternative pathway exists, known as cross-presentation, involving the uptake of extracellular antigens, their retrograde transfer from phagosomes to the cytoplasm, and subsequent degradation for MHC-I loading in proteasomes and the ER.
▲Neoantigen production and presentation pathways[3]
Humans have over 24,000 different human leukocyte antigen class I (HLA-I, including HLA-A, -B, and -C) and class II (HLA-DR, HLA-DQ, and HLA-DP) alleles, resulting in a diverse polymorphism. The combination of these alleles contributes to the diversity of polymorphism. The patient's HLA alleles determine their tumor-specific neoantigen repertoire, which is presented for recognition by T cells. Additionally, HLA loss of heterozygosity (HLA-LOH), occurring in 40% of non-small cell lung cancers, impairs neoantigen presentation, promoting immune evasion. Therefore, one of the crucial initial steps in neoantigen prediction is identifying the patient's HLA genotype. Various computational methods can now be applied to Next-Generation Sequencing (NGS) data to achieve this goal.
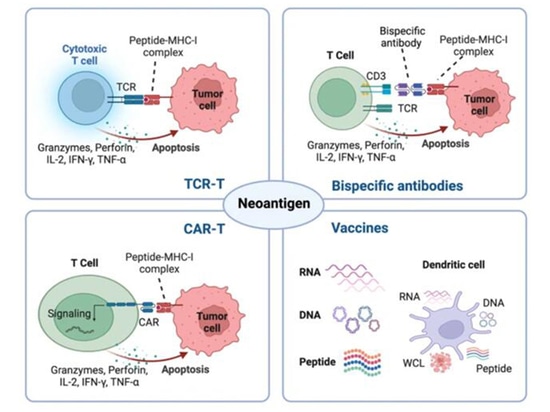
Therapeutic Strategies Based on Neoantigens
Due to the absence of thymic selection and central tolerance, tumor-specific neoantigens generated by genetic alterations elicit high-affinity T cells. Leveraging their tumor specificity and immunogenicity, neoantigens serve as emerging targets for cancer immunotherapy, including tumor vaccines, adoptive cell therapies (ACT), antibody-based treatments, and potential predictors for immune checkpoint inhibitors (ICBs) [3].
▲Molecular characteristics of diffuse high-grade glioma in children[3]
The new antigen is composed of personalized new antigens specifically targeting each patient or common new antigens expressed in many cancer patients. Therapies based on common new antigens are more resource and time-efficient than personalized new antigen therapies. Because personalized new antigens are patient-specific, they cannot be used to target a large number of patients. With the latest advances in high-throughput sequencing, personalized new antigens enable the immune system to target immunogenic epitopes on malignant tumors without predefined common antigens.
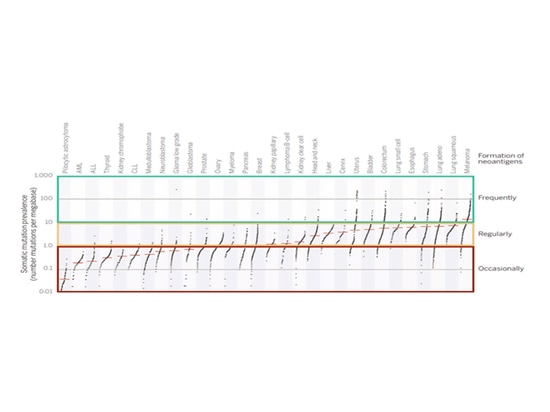
Relationship between TMB and TNB
In most melanoma patients, TMB is greater than 10, leading to the generation of more effective neoantigens. Based on these data, it can be predicted that if TMB is greater than 10 in the tumor, a considerable number of neoantigens will be produced. If TMB is between 1 and 10, there is still a possibility of carrying neoantigens. Generally, if TMB is less than 1 in the tumor, it is often difficult to generate neoantigens recognizable by T cells. Tumors with high TMB (>10) indicate that there are more tumor neoantigens on the surface of tumor cells, resulting in more effective killing by immune cells. Additionally, patients with high TMB may exhibit better treatment responses to immune checkpoint inhibitors.
▲Tumor TMB and the potential to produce neoantigens[4]
The Challenges of Clinical Application
1. Immunotherapy based on new antigens shows objective efficacy only in a small subset of well-documented patient responses. Therefore, significant improvements are needed to enhance clinical outcomes, including increasing the accuracy of predicting new antigens, overcoming immune evasion, and optimizing the pipeline for the production process.
2. Limited Accuracy of New Antigen Prediction: The widespread application of personalized immunotherapy is restricted by the limited discovery of cancer-specific new antigens, due to the heterogeneity of mutation loads and significant differences in the presentation of new antigens among different tumor types. Only 10% of non-synonymous tumor cell mutations can generate mutated peptides with high MHC affinity, and only 1% of MHC-bound peptides are recognized by patient T cells.
3. Loss of New Antigens: The absence of tumor-specific new antigens may be a crucial immune evasion strategy for tumors. The loss of new antigens can be induced through various pathways, such as copy number loss, transcriptional suppression, epigenetic silencing, and post-translational mechanisms.
4. Insufficient Production of New Antigen-Specific T Cells: The Foshu Bioscience Panorama 602 gene detection product includes HLA-I heterozygosity loss detection for assessing the benefits of adjuvant immunotherapy. It also includes information on targeted therapies, chemotherapy, tumor mutational burden (TMB), tumor neoantigen burden (TNB), immune positive and negative factors, and other relevant information on drug use, subtyping, prognosis, genetics, etc., to provide patients with the maximum possible benefit.
References
1. Nat Rev Cancer. 2017 Apr;17(4):209-222.
2.Viral Immunol. 2020 Apr;33(3):160-178.
3.Signal Transduct Target Ther. 2023 Jan 6;8(1):9.
4.Science. 2015 Apr 3;348(6230):69-74.
Statement: This article is for sharing only. If there are any copyright issues, please contact us as soon as possible and we will correct it as soon as possible. Thank you!